

Make 2x the Impact Now
Make 2x the Impact Now
Our March Mission Match is underway, but not for long. Your gift by March 10 can go twice as far to advance research and help provide care and support for those living with Alzheimer’s and their caregivers.
Give NowCreutzfeldt-Jakob Disease
Creutzfeldt-Jakob disease (CJD) is the most common human form of a group of rare, fatal brain disorders known as prion diseases.
About Creutzfeldt-Jakob disease
 Prion diseases, such as Creutzfeldt-Jakob disease, occur when prion protein, which is found throughout the body but whose normal function isn't yet known, begins folding into an abnormal three-dimensional shape. This shape change gradually triggers prion protein in the brain to fold into the same abnormal shape.
Prion diseases, such as Creutzfeldt-Jakob disease, occur when prion protein, which is found throughout the body but whose normal function isn't yet known, begins folding into an abnormal three-dimensional shape. This shape change gradually triggers prion protein in the brain to fold into the same abnormal shape.
Creutzfeldt-Jakob disease causes a type of dementia that gets worse unusually fast. More common causes of dementia, such as Alzheimer's, dementia with Lewy bodies and frontotemporal dementia, typically progress more slowly.
Through a process scientists don't yet understand, misfolded prion protein destroys brain cells. Resulting damage leads to rapid decline in thinking and reasoning as well as involuntary muscle movements, confusion, difficulty walking and mood changes.
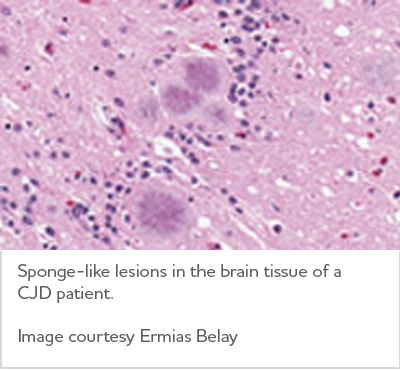 Creutzfeldt-Jakob disease is rare, occurring in about one in 1 million people annually worldwide.
Creutzfeldt-Jakob disease is rare, occurring in about one in 1 million people annually worldwide.
Experts generally recognize the following main types of Creutzfeldt-Jakob disease:
- Sporadic Creutzfeldt-Jakob disease develops spontaneously for no known reason. It accounts for 85% of cases. On average, sporadic Creutzfeldt-Jakob disease first appears between ages 60 and 65.
- Familial Creutzfeldt-Jakob disease is caused by certain changes in the chromosome 20 gene coding the biological blueprint for prion protein. People who develop familial Creutzfeldt-Jakob disease do so because they inherited the genetic changes from a parent. Familial Creutzfeldt-Jakob disease accounts for about 10-15% percent of cases. It develops, on average, at a younger age than sporadic Creutzfeldt-Jakob disease, with some genetic types appearing as early as ages 20 to 40.
- Acquired Creutzfeldt-Jakob disease results from exposure to an external source of abnormal prion protein. These sources are estimated to account for about 1% of Creutzfeldt-Jakob disease cases. The two most common outside sources are:
- Medical procedures involving instruments used in neurosurgery, growth hormone from human sources or certain transplanted human tissues, including corneas (the clear outer covering of the eye) and dura mater (the fibrous membrane covering the brain and spinal cord). This type of acquired CJD is also known as iatrogenic CJD (iCJD). The risk of iCJD from medical procedures has been greatly reduced by improved neurosurgical instruments sterilization techniques, new single-use instruments and synthetic sources of growth hormone and dura mater.
- Meat or other products from cattle infected with bovine spongiform encephalopathy (BVE) or "mad cow disease," recognized in the mid-1990s as the cause of variant CJD (vCJD). Scientists traced this new type of CJD to consumption of beef from cattle whose feed included processed brain tissue from other animals. Since then, experts have diagnosed about 200 cases of vCJD, primarily in the United Kingdom and other European countries. Variant CJD tends to occur at a younger age than sporadic or familial forms, sometimes even in teenagers. New cases of vCJD have slowed significantly, most likely due to changes in animal feeding practices.
Chronic wasting disease
This prion disease is similar to mad cow disease that's been found in wild deer, elk and moose. According to the Centers for Disease Control (CDC), there's no evidence to date that the disease has been transmitted to humans.
Symptoms
Specific Creutzfeldt-Jakob disease symptoms experienced by an individual and the order in which they appear can differ significantly. Some common symptoms include:
- Depression.
- Agitation, apathy and mood swings.
- Rapidly worsening confusion.
- Disorientation.
- Problems with memory, thinking, planning and judgment.
- Difficulty walking.
- Muscle stiffness, twitches and involuntary jerky movements.
- Vision problems, such as double vision and hallucinations
Diagnosis
Rapid symptom progression is one of the most important clues that a person may have Creutzfeldt-Jakob disease.
There is no single test — or any combination of tests — that can conclusively diagnose sporadic Creutzfeldt-Jakob disease in a living person, but the following tests may help determine whether an individual has Creutzfeldt-Jakob disease:
- Electroencephalogram (EEG) measures the brain's patterns of electrical activity similar to the way an electrocardiogram (ECG) measures the heart's electrical activity.
- Brain magnetic resonance imaging (MRI) can detect certain brain changes consistent with Creutzfeldt-Jakob disease.
- Lumbar puncture (spinal tap) tests spinal fluid for the presence of certain proteins.
- Protein misfolding cyclic amplification (PMCA): PMCA is an amplification
technique for the detection of misfolded protein aggregates.
Causes and risks
Sporadic CJD has no known cause. Most scientists believe the disease begins when prion protein somewhere in the brain spontaneously misfolds, triggering a "domino effect" that misfolds prion protein throughout the brain. Genetic variation in the prion protein gene at a location called "codon 129" may increase risk of this spontaneous misfolding.
Variation at codon 129 in the prion protein gene may also play a role in making people susceptible to acquired CJD from external sources. Scientists don’t yet know why acquired CJD seems to be transmitted through such a limited number of external sources. Researchers have found no evidence that the abnormal protein is commonly transmitted through sexual activity or blood transfusions, although a few cases of vCJD seem to have been spread through blood transfusions. Professionals who regularly encounter blood from a human or animal, such as surgeons, pathologists or butchers, have not been shown to have a higher-than-normal risk through occupational exposure.
Familial CJD is caused by variations in the prion protein gene that increase the likelihood an individual will develop CJD. Researchers have identified more than 50 prion protein mutations in those with inherited CJD. Genetic testing can determine whether family members at risk have inherited a CJD-causing mutation. Experts strongly recommend professional genetic counseling both before and after genetic testing for hereditary CJD.
Age has an influence on sporadic CJD, which tends to develop later in life, usually around age 60. Onset of familial CJD occurs slightly earlier and vCJD has affected people at a much younger age, usually in their late 20s.
Chronic wasting disease is a prion disease similar to mad cow disease that’s been found in wild deer, elk and moose in certain U.S. states, Canadian provinces, Korea and Norway. According to the U.S. Centers for Disease Control and Prevention (CDC), there’s no evidence to date that chronic wasting disease has been transmitted to humans, including hunters who eat meat from affected animals. There’s also no evidence that rates of CJD have increased in states or provinces where chronic wasting disease has been identified. Additional studies are under way to understand what risk, if any, chronic wasting disease poses to humans. The CDC recommends that hunters who plan to eat meat from deer, elk or moose in areas where chronic wasting disease occurs consider having the meat tested by their local state wildlife agency. The CDC also recommends wearing gloves while field dressing these animals to avoid handling the brain or spinal column.
Treatment and outcomes
There is no treatment that can slow or stop the underlying brain cell destruction caused by Creutzfeldt-Jakob disease and other prion diseases. Various drugs have been tested but have not shown any benefit. Clinical studies of potential Creutzfeldt-Jakob disease treatments are complicated by the rarity of the disease and its rapid progression.
Current therapies focus on treating symptoms and on supporting individuals and families coping with Creutzfeldt-Jakob disease. Doctors may prescribe painkillers such as opiates to treat pain if it occurs. Muscle stiffness and twitching may be treated with muscle-relaxing medications or antiseizure drugs. In the later stages of the disease, individuals with Creutzfeldt-Jakob disease become completely dependent on others for their daily needs and comfort.
Creutzfeldt-Jakob disease progresses rapidly. Those affected lose their ability to move or speak and require full-time care to meet their daily needs. An estimated 90 percent of those diagnosed with sporadic Creutzfeldt-Jakob disease die within one year. Those affected by familial Creutzfeldt-Jakob disease tend to develop the disorder at an earlier age and survive somewhat longer than those with the sporadic form, as do those diagnosed with variant Creutzfeldt-Jakob disease. Scientists have not yet learned the reason for these differences in survival.
Help is available
Creutzfeldt-Jakob Disease Foundation is a nonprofit organization that offers support, information and guidance to those dealing with Creutzfeldt-Jakob disease. Call the Foundation at 800.659.1991.
The Alzheimer's Association can help you learn more about Alzheimer's and other dementias, and help you find local support services. Call our 24/7 Helpline at 800.272.3900.
Social Security Administration (SSA) has a "compassionate allowance" program in which workers diagnosed with Creutzfeldt-Jakob disease can qualify for Social Security disability benefits. Call the SSA at 800.772.1213.
Medline Plus: Creutzfeldt-Jakob disease is a consumer health information service of the U.S. National Library of Medicine and National Institutes of Health (NIH). This gateway page links to resources from NIH agencies, major medical centers and other sources.
Related Pages

The Alzheimer’s Association is in your community.
Find Your Local Chapter
Learn how Alzheimer’s disease affects the brain.
Take the Brain Tour